EU Pharmaceutical Package: Key Changes for the Life Sciences Industry
Update Health Care & Life Sciences 3/2026
Shortly before the turn of the year, the EU institutions reached agreement on a comprehensive reform of European pharmaceutical law. Following six months of intensive trilogue negotiations, it remained uncertain until the very end whether consensus could be achieved on the most controversial issues. The outcome addresses the core objectives of the reform: improved access to medicinal products, strengthening innovation and competitiveness, security of supply, combating antimicrobial resistance and more efficient procedures.
With the political agreement, the legislative process has entered its final phase. In early March, the provisionally final drafts of the new Regulation (“Draft Regulation”) and the new Directive (“Draft Directive”) were published. No material changes are expected at this stage and companies should therefore begin preparing for the foreseeable changes now.
I. Regulatory Protection Periods: Data Exclusivity and Market Protection
The regulation of protection periods was among the most controversial aspects of the reform. After long negotiations, the Commission, Parliament and Council agreed on a base data exclusivity period of eight (8) years (Art. 80(1) Draft Directive), thereby maintaining the current level of protection. During this period, competitors may not rely on the pre-clinical and clinical study data and other documentation of the marketing authorisation holder in order to obtain a marketing authorisation through the simplified procedure. An extension of this data exclusivity period is generally not contemplated. The sole exception is a one-year extension through the Transferable Exclusivity Voucher – on which more below.
Following data exclusivity, a one (1) year market protection period will apply going forward, one year less than under the current regime (Art. 80(2) Draft Directive). During this period, competing products such as generics and biosimilars whose marketing authorisations reference the data of the reference medicinal product may not be marketed. Market protection may be extended by twelve (12) months if the medicinal product falls within one of the following four categories:
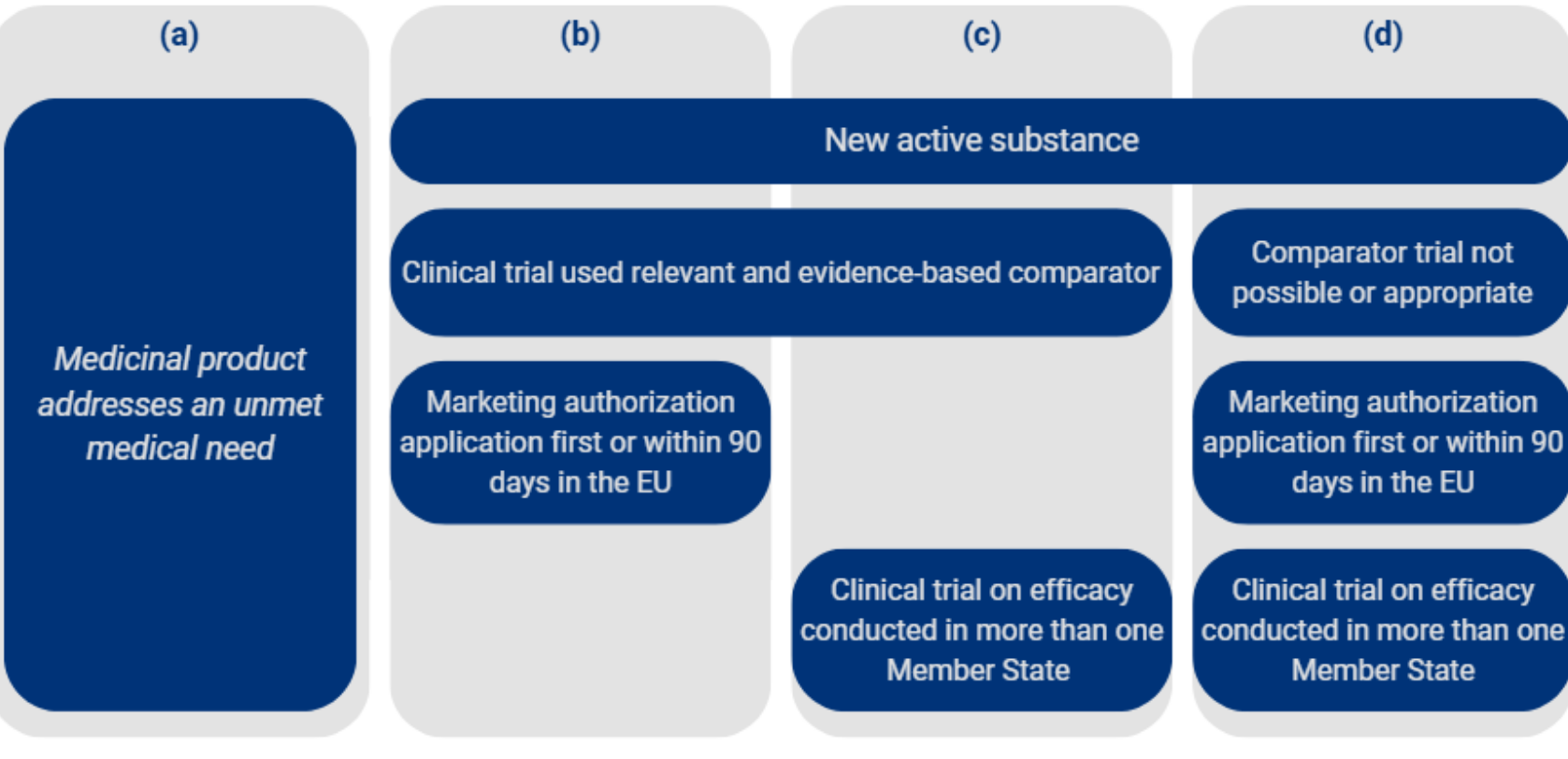
A further one-year extension of market protection is available where the marketing authorisation holder obtains, during the data exclusivity period, an authorisation for one or more additional therapeutic indications for which a significant clinical benefit compared to existing therapies has been demonstrated (Art. 81(2a) Draft Directive). This extension may expressly be granted only once. The maximum duration of market protection is capped at two (2) years, unless an extension for an additional indication is granted (Art. 81(2b) Draft Directive). In that case, a total of three (3) years of market protection is possible. The overall protection period is thus limited to a total of eleven (11) years, in line with the current legal position. From the perspective of research-based pharmaceutical companies, it is a positive development that the originally proposed significant reduction of data exclusivity has been taken off the table and the baseline protection of eight years has been preserved. At the same time, planning uncertainty for pharmaceutical companies is increasing: the complex conditions attached to the market protection extension options are only partially within a company’s control.
It should also be noted that market protection may cease to apply in individual Member States where the marketing authorisation holder has failed to comply with a request from the relevant Member State to make the medicinal product available in sufficient quantities in that country (Art. 56a Draft Directive, on this in more detail under VI.).
Unsurprisingly, the concept of so-called drug repurposing has also been included in the final version of the new Directive. Under this provision, medicinal products authorised for a new therapeutic indication are granted a one-time data exclusivity period of four (4) years (Art. 84 Draft Directive), provided that no data exclusivity previously existed for the product or the initial authorisation was granted at least 25 years ago. This creates an incentive both for research involving known active substances and for the swift and cost-effective provision of new medicinal products.
II. Bolar Exemption
The reform clarifies and broadens the so-called Bolar exemption in order to enable generic manufacturers to enter the market immediately upon expiry of patent protection. The agreement clarifies that neither patents nor rights arising from a supplementary protection certificate are infringed where studies, trials and other activities necessary for obtaining a marketing authorisation are conducted (Art. 85(1) Draft Directive). The scope extends to health technology assessments (HTA), pricing and reimbursement decisions, as well as public procurement procedures. In addition, it has been clarified that intellectual property rights of the reference medicinal product do not constitute a valid ground for refusing, revoking or suspending decisions taken within the scope of the Bolar exemption (Art. 85(2) Draft Directive).
This new provision is likely to contribute to the harmonisation of the Bolar exemption across the EU. At the same time, concerns have been raised regarding potential adverse effects on the competitiveness of the European pharmaceutical industry.
III. Orphan Drugs
A central objective of the reform is to promote research and development of medicinal products for rare diseases – so-called orphan drugs. The concept of “high unmet medical need”, originally proposed by the Commission but subsequently deleted, has been reintroduced under the new designation “breakthrough orphan medicinal product”. Under Art. 70 of the Draft Regulation, this category encompasses drugs whose use results in a clinically relevant reduction in disease morbidity or mortality in the relevant patient population, provided that no authorised medicinal product for the rare condition in question exists in the Union.
Market exclusivity is governed on a differentiated basis: for standard orphan drugs, an exclusivity period of nine (9) years is provided for, whereas breakthrough orphan drugs will enjoy an extended exclusivity period of eleven (11) years. Where the authorisation is based on bibliographical data, market exclusivity is limited to four (4) years.
In addition, market exclusivity may be extended twice by twelve (12) months each. The prerequisite is that the marketing authorisation holder obtains, at least two years before the expiry of the exclusivity period, an authorisation for one or more new therapeutic indications for a different rare disease. This provision represents a significant departure from the existing legal framework, under which market exclusivity could be obtained separately and in full for each therapeutic indication for a rare disease.
IV. Antimicrobials
To effectively face the growing threat of antimicrobial resistance, the reform introduces an innovative voucher system: the so-called Transferable Exclusivity Vouchers (TEVs). These vouchers extend data exclusivity for a priority antimicrobial by twelve months (Art. 40 Draft Regulation). The system is designed with flexibility: companies may apply the TEV either to the priority antimicrobial itself or to another centrally authorised medicinal product of the same marketing authorisation holder. Moreover, transfer to third parties is possible. Where the TEV is used for a different medicinal product, it may be exercised in the fifth or sixth year of the base data exclusivity period. This represents an expansion compared to the original proposal, which provided for use only in the fifth year. However, a so-called blockbuster restriction applies. Vouchers may not be used for medicinal products whose gross annual turnover in the EU exceeds EUR 490 million within the first four years following authorisation. As this constitutes a novel incentive mechanism, the system is initially limited to 15 years. The Commission may issue a maximum of five vouchers during this period (Art. 43 Draft Regulation).
In addition, the reform confirms the subscription model (“Netflix” model) introduced by Parliament as a voluntary procurement mechanism for antimicrobials (Art. 43a Draft Regulation). Member States may jointly enter into multi-year subscription contracts under which remuneration is delinked from sales volumes. The company receives an agreed regular payment irrespective of prescription and sales figures and in return undertakes to ensure a continuous and sufficient supply of defined quantities.
Furthermore, the reform strengthens stewardship measures. A mandatory prescription requirement for antimicrobial agents is to apply EU-wide (Art. 51 Draft Directive). Marketing authorisation applications for antimicrobials must include a stewardship plan (Art. 17 Draft Directive) and the risk of antimicrobial resistance must be taken into account in the environmental risk assessment (Art. 22(4) Draft Directive).
This package of measures is intended to provide predictable revenues for the refinancing of research and development of antimicrobials. At the same time, it promotes a restrained, needs-based use of new antimicrobials, thereby aiming to contain the development of resistance.
V. Acceleration and Digitalisation
The reform contains important elements aimed at accelerating and modernising procedures. As a result of the trilogue negotiations, the standard review and authorisation period under the centralised procedure at the EMA will be shortened from the current 210 to 180 days – the Commission’s draft proposal has prevailed on this point. Industry has welcomed this acceleration of the approval processes.
Also to be viewed positively are the introduction of regulatory sandboxes and the further digitalisation of procedures. Applications must in future be submitted uniformly in electronic form and package leaflets must be made available in digital format. To reduce administrative burden, marketing authorisations are in principle to be granted for an unlimited duration. The EMA may derogate from this on safety grounds.
VI. New Obligations: Security of Supply and Shortage Management
The supply obligations introduced by the Council have become part of the reform and may affect centrally authorised medicinal products (Art. 5a Draft Regulation). Member States may request a marketing authorisation holder to make its medicinal product available on the respective market in sufficient quantities. To this end, they may either use their own procedure or rely on the mechanism under the new Art. 56a Draft Directive. Marketing authorisation holders concerned are generally obliged to comply with such a request, unless exceptional, unforeseeable or demonstrably uncontrollable circumstances exist. Non-compliance does not, for the time being, entail financial sanctions, contrary to earlier proposals by Parliament. An evaluation by the Commission in four years’ time is to determine whether sanctioning mechanisms for this obligation should be introduced.
Where data exclusivity or market exclusivity exists for a medicinal product, Art. 56a Draft Directive additionally provides the following consequences: if a marketing authorisation holder fails to comply with a Member State’s request within three years, the market protection under Art. 80(2) Draft Directive and, in the case of orphan drugs, the extension of market exclusivity for new therapeutic indications, shall not apply in that Member State. Generics and biosimilars may then be placed on the market in that Member State, even though market protection continues to apply in other Member States. To ensure that the end of market protection in one Member State is not abused to effectively circumvent protection in other Member States, wholesaler and distance sale distributors are prohibited from placing such medicinal products on markets where the protection of the originator product is still in force (Art. 166(5) Draft Directive). Additionally, under certain conditions, marketing authorisation applications for generics and biosimilars may be validated and assessed as early as six years after the commencement of data exclusivity for the reference medicinal product. The authorisation itself, however, may only be granted upon expiry of the full eight-year protection period.
The obligations relating to the prevention of supply shortages are being expanded. A general obligation to prepare Shortage Prevention Plans (SPPs) applicable to all medicinal products has not been adopted. However, such an obligation applies to all prescription-only medicinal products (Art. 117 Draft Regulation). Companies must report imminent shortages at least six months in advance (Art. 116 Draft Regulation). For small and medium-sized companies in particular, this is likely to entail a significant additional burden.
VII. Next Steps
On 18 March, the Committee on Public Health (SANT) in Parliament voted positively on the outcome of the trilogue negotiations. This paves the way for translation of the legislative texts into all Union languages and formal endorsement by the Council and Parliament, which could take place as early as this autumn.
The new Regulation enters into force 20 days after publication in the Official Journal of the EU, with application commencing generally 24 months after entry into force. Immediate application or a shorter transitional period is foreseen only for a limited number of exceptions, including the provisions on orphan drugs, TEVs and regulatory sandboxes. The new Directive is subject to a transposition period of 24 months. In addition, delegated acts and guidelines are expected. These will have a significant influence on whether the reform ultimately achieves the objectives it pursues.