EU-Pharmapaket: Die wichtigsten Änderungen für die Life-Sciences-Industrie im Überblick
Update Health Care & Life Sciences 3/2026
Kurz vor dem Jahreswechsel haben sich die EU-Institutionen auf eine umfassende Reform des europäischen Arzneimittelrechts geeinigt. Nach einem halben Jahr intensiver Trilog-Verhandlungen war bis zuletzt unklar, ob bei den besonders strittigen Punkten ein Konsens gelingen würde. Das Ergebnis adressiert die Kernziele der Reform: besserer Zugang zu Arzneimitteln, Stärkung von Innovation und Wettbewerbsfähigkeit, Versorgungssicherheit, Bekämpfung antimikrobieller Resistenzen sowie effizientere Verfahren. Mit der politischen Einigung ist das Gesetzgebungsverfahren in seine Schlussphase eingetreten. Anfang März wurden die vorläufig finalen Entwürfe der neuen Verordnung („VO-E“) und der neuen Richtlinie („RL-E“) veröffentlicht. Wesentliche Änderungen sind nicht mehr zu erwarten, Unternehmen sollten sich daher jetzt auf die absehbaren Neuerungen einstellen.
I. Schutzfristen: Unterlagenschutz und Vermarktungsschutz
Die Regelung der Schutzfristen gehörte zu den strittigsten Aspekten der Reform. Nach langem Ringen haben sich Kommission, Parlament und Rat auf einen grundlegenden Unterlagenschutz von acht (8) Jahren geeinigt, Art. 80 Abs. 1 RL-E, dies entspricht dem Status quo. In dieser Zeit dürfen Wettbewerber auf die präklinischen und klinischen Studiendaten und weiteren Unterlagen des Zulassungsinhabers nicht Bezug nehmen, um eine Zulassung im vereinfachten Verfahren zu erhalten. Eine Verlängerung dieser Schutzfrist ist grundsätzlich nicht vorgesehen. Die einzige Ausnahme bildet die einjährige Verlängerung durch den Transferable Exclusivity Voucher – dazu später mehr.
An den Unterlagenschutz schließt sich künftig ein einjähriger (1) Vermarktungsschutz an und damit ein Jahr weniger als bisher, Art. 80 Abs. 2 RL-E. In dieser Zeit dürfen Wettbewerbsprodukte wie Generika und Biosimilars, deren Zulassungen auf die Unterlagen des Referenzarzneimittels Bezug nehmen, nicht vermarktet werden. Der Vermarktungsschutz kann um zwölf (12) Monate verlängert werden, wenn das Arzneimittel in eine der folgenden vier Kategorien fällt:
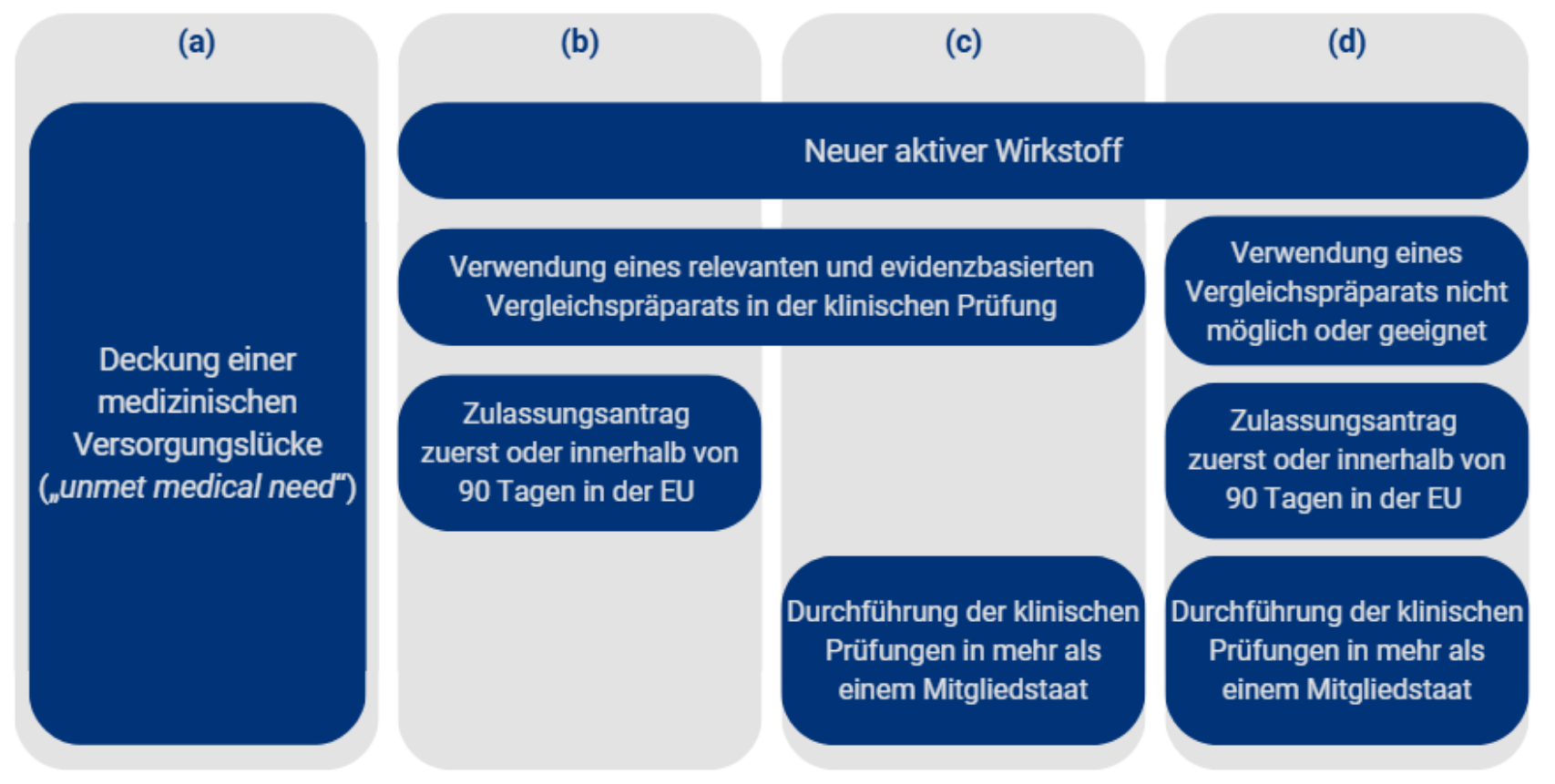
Eine zusätzliche Verlängerung des Vermarktungsschutzes um ein weiteres Jahr ist möglich, wenn der Zulassungsinhaber während der Unterlagenschutzfrist eine Zulassung für eine oder mehrere zusätzliche therapeutische Indikationen erhält, für die ein signifikanter klinischer Nutzen im Vergleich zu bestehenden Therapien nachgewiesen ist (Art. 81 Abs. 2a RL-E). Diese Verlängerung kann ausdrücklich nur einmal gewährt werden. Die maximale Dauer des Vermarktungsschutzes ist auf zwei (2) Jahre begrenzt, es sei denn, es wird eine Verlängerung für eine Zusatzindikation gewährt, Art. 81 Abs. 2b RL-E. In diesem Fall sind insgesamt drei (3) Jahre Vermarktungsschutz möglich. Damit ist die Schutzfrist auf insgesamt elf (11) Jahre begrenzt, entsprechend der aktuellen Rechtslage. Positiv ist aus Sicht der forschenden Arzneimittelhersteller, dass die ursprünglich geplante deutliche Kürzung des Unterlagenschutzes vom Tisch ist und der Basisschutz von acht Jahren erhalten bleibt. Zugleich wächst die Planungsunsicherheit für pharmazeutische Unternehmen: Die komplexen Bedingungen für die Verlängerungsoptionen des Vermarktungsschutzes sind nur teilweise steuerbar.
Zu beachten ist ferner: Der Vermarktungsschutz kann in einzelnen Mitgliedstaaten entfallen, wenn der Zulassungsinhaber einer Aufforderung des jeweiligen Mitgliedstaats, das Arzneimittel dort in ausreichender Menge zur Verfügung zu stellen, nicht nachgekommen ist (Art. 56a RL-E, dazu näher unter VI.).
Wenig überraschend findet sich auch das sogenannte Drug Repurposing in der finalen Fassung der neuen Richtlinie. Dabei wird Arzneimitteln für die Zulassung einer neuen therapeutischen Indikation einmalig ein Unterlagenschutz von vier (4) Jahren gewährt (Art. 84 RL-E), wenn für das Arzneimittel bislang kein Unterlagenschutz bestand oder die Erstzulassung mindestens 25 Jahre zurückliegt. Damit wird ein Anreiz sowohl für die Forschung mit bereits bekannten Wirkstoffen als auch für die schnelle und kostengünstige Bereitstellung neuer Arzneimittel geschaffen.
II. Bolar-Ausnahme
Die Reform präzisiert und erweitert die sogenannte Bolar‑Ausnahme, um Generikaanbietern den Markteintritt unmittelbar nach Ablauf des Patentschutzes zu ermöglichen. Die Einigung stellt klar, dass weder Patente noch Rechte aus einem ergänzenden Schutzzertifikat verletzt werden, sofern erforderliche Studien, Prüfungen und sonstige Tätigkeiten zur Erlangung einer Marktzulassung durchgeführt werden (Art. 85 Abs. 1 RL-E). Der Anwendungsbereich erstreckt sich auf Health-Technology-Assessments (HTA), Preis- und Erstattungsentscheidungen sowie Vergabeverfahren. Ergänzend wurde klargestellt, dass Rechte des geistigen Eigentums am Referenzarzneimittel keinen zulässigen Grund darstellen, um Entscheidungen im Rahmen der Bolar-Ausnahme abzulehnen, zu widerrufen oder auszusetzen (Art. 85 Abs. 2 RL-E).
Diese Neuregelung dürfte zu einer Harmonisierung der Bolar-Ausnahme innerhalb der EU beitragen. Zugleich werden Bedenken hinsichtlich möglicher negativer Auswirkungen auf die Wettbewerbsfähigkeit der europäischen Pharmaindustrie geäußert.
III. Orphan Drugs
Ein zentrales Anliegen der Reform ist die Förderung von Forschung und Entwicklung bei Arzneimitteln gegen seltene Erkrankungen – den sogenannten Orphan Drugs. Das zunächst von der Kommission vorgeschlagene, dann aber gestrichene Konzept des „hohen ungedeckten medizinischen Bedarfs“ wurde unter der neuen Bezeichnung „Breakthrough-Orphan Drug“ wieder aufgegriffen. Hierunter fallen gemäß Art. 70 VO-E Arzneimittel, deren Anwendung zu einer klinisch relevanten Verringerung der Morbidität oder Mortalität der betreffenden Patientengruppe führt, sofern in der Union kein zugelassenes Arzneimittel gegen das jeweilige seltene Leiden existiert.
Die Marktexklusivität wird differenziert geregelt: Für reguläre Orphan Drugs ist künftig eine Exklusivitätsfrist von neun (9) Jahren vorgesehen, während Breakthrough-Orphan Drugs eine verlängerte Exklusivität von elf (11) Jahren genießen. Erfolgt die Zulassung auf Grundlage bibliografischer Daten, beträgt die Marktexklusivität lediglich vier (4) Jahre.
Darüber hinaus kann die Marktexklusivität zweimal um jeweils zwölf (12) Monate verlängert werden. Voraussetzung ist, dass der Zulassungsinhaber mindestens zwei Jahre vor Ablauf der Exklusivitätsfrist eine Zulassung für eine oder mehrere neue therapeutische Indikationen für eine andere seltene Erkrankung erhält. Diese Regelung beinhaltet eine signifikante Abweichung von der bisherigen Rechtslage, nach der die Marktexklusivität gesondert und in vollem Umfang für jede therapeutische Indikation für eine seltene Erkrankung erlangt werden kann.
IV. Antimikrobielle Arzneimittel
Um der wachsenden Bedrohung durch antimikrobielle Resistenzen wirksam zu begegnen, führt die Reform ein innovatives Gutscheinsystem ein: die sogenannten Transferable Exclusivity Vouchers (TEV). Diese Gutscheine verlängern den Unterlagenschutz für ein prioritäres Antibiotikum um zwölf Monate (Art. 40 VO-E). Das System ist flexibel ausgestaltet: Unternehmen können den TEV entweder auf das prioritäre Antibiotikum selbst oder auf ein anderes zentral zugelassenes Arzneimittel desselben Zulassungsinhabers anwenden.
Darüber hinaus ist eine Übertragung auf Dritte möglich. Wird der TEV für ein anderes Arzneimittel genutzt, kann er im fünften oder sechsten Jahr des regulären Unterlagenschutzes eingesetzt werden. Dies stellt eine Erweiterung gegenüber dem ursprünglichen Vorschlag dar, der nur das fünfte Jahr vorsah. Allerdings gilt eine sogenannte Blockbuster-Beschränkung: Gutscheine dürfen nicht für Arzneimittel eingesetzt werden, deren Bruttojahresumsatz in der EU innerhalb der ersten vier Jahre nach Zulassung 490 Millionen Euro übersteigt. Da es sich um einen neuartigen Anreizmechanismus handelt, ist das System zunächst auf 15 Jahre begrenzt. Die Kommission kann in diesem Zeitraum maximal fünf Gutscheine erteilen (Art. 43 VO-E).
Ergänzend bestätigt die Reform das vom Parlament eingebrachte Abonnement‑Modell („Netflix“-Modell) als freiwilligen Beschaffungsmechanismus für antimikrobielle Arzneimittel (Art. 43a VO-E). Mitgliedstaaten können gemeinsam mehrjährige Abonnementverträge abschließen, bei denen die Vergütung vom Absatzvolumen entkoppelt wird (Delinkage-Prinzip). Das Unternehmen erhält eine vereinbarte regelmäßige Zahlung, unabhängig von Verordnungs- und Verkaufszahlen, und verpflichtet sich im Gegenzug zu einer kontinuierlichen und ausreichenden Belieferung mit definierten Mengen.
Darüber hinaus verschärft die Reform die Stewardship‑Maßnahmen. EU‑weit soll künftig eine verpflichtende ärztliche Verschreibungspflicht für antimikrobielle Wirkstoffe gelten (Art. 51 RL-E). Zulassungsanträge für antimikrobielle Arzneimittel müssen einen Stewardship‑Plan enthalten (Art. 17 RL-E), und das Risiko antimikrobieller Resistenzen ist in der Umweltrisikobewertung zu berücksichtigen (Art. 22 Abs. 4 RL-E).
Dieses Maßnahmenpaket soll planbare Einnahmen zur Refinanzierung von Forschung und Entwicklung antimikrobieller Arzneimittel ermöglichen. Gleichzeitig fördert es eine zurückhaltende, bedarfsorientierte Nutzung neuer Antibiotika und soll so die Resistenzentwicklung eindämmen.
V. Beschleunigung und Digitalisierung
Die Reform enthält wichtige Elemente zur Beschleunigung und Modernisierung der Verfahren. Als Ergebnis der Trilog-Verhandlungen wird die reguläre Prüf- und Zulassungsdauer im zentralisierten Verfahren bei der EMA von bislang 210 auf 180 Tage verkürzt – damit setzt sich die Linie des Kommissionsentwurfs durch. Die Industrie begrüßt diese Beschleunigung der Genehmigungsprozesse.
Positiv zu bewerten sind auch die Einführung von Regulatory Sandboxes sowie die weitergehende Digitalisierung der Verfahren. Anträge sind künftig einheitlich elektronisch einzureichen und Packungsbeilagen müssen in digitaler Form bereitgestellt werden. Zur Reduzierung administrativer Lasten sollen Marktzulassungen grundsätzlich unbefristet gelten. Die EMA kann hiervon aus Sicherheitsgründen abweichen.
VI. Neue Pflichten: Versorgungssicherheit und Engpassmanagement
Die vom Rat eingebrachten Lieferverpflichtungen sind Teil der Reform geworden und können zentral zugelassene Arzneimittel betreffen, Art. 5a VO-E. Mitgliedstaaten können einen Zulassungsinhaber auffordern, sein Arzneimittel auf dem jeweiligen Markt in ausreichender Menge zur Verfügung zu stellen. Hierzu können sie entweder ein eigenes Verfahren nutzen oder auf den Mechanismus des neuen Art. 56a RL-E zurückgreifen. Betroffene Zulassungsinhaber sind grundsätzlich verpflichtet, dieser Aufforderung nachzukommen, es sei denn, es bestehen außergewöhnliche, unvorhersehbare oder nachweislich unkontrollierbare Umstände. Eine Nichteinhaltung zieht vorerst keine finanziellen Sanktionen nach sich, anders als zuvor vom Parlament vorgeschlagen. Eine Evaluierung der Kommission in vier Jahren soll bestimmen, ob eine Sanktionierung dieser Verpflichtung ergänzt werden sollte.
Soweit ein Unterlagenschutz oder eine Marktexklusivität für ein Arzneimittel besteht, regelt Art. 56a RL-E darüber hinaus folgende Konsequenzen: Kommt ein Zulassungsinhaber der Aufforderung eines Mitgliedstaates innerhalb von drei Jahren nicht nach, so findet der Vermarktungsschutz nach Art. 80 Abs. 2 RL-E und bei Orphan Drugs die Verlängerung der Marktexklusivität für neue therapeutische Indikationen in diesem Mitgliedstaat keine Anwendung. Generika und Biosimilars können dort auf den Markt gebracht werden, obwohl in anderen Mitgliedstaaten noch Vermarktungsschutz besteht. Um sicherzustellen, dass der Wegfall des Vermarktungsschutzes in einem Mitgliedstaat nicht dazu missbraucht wird, den Schutz in anderen Mitgliedstaaten faktisch zu unterlaufen, ist es Großhändlern und dem Fernabsatz untersagt, diese Arzneimittel in Märkte zu bringen, in denen der Schutz des Originalpräparats noch läuft (Art. 166 Abs. 5 RL-E). Außerdem wird unter bestimmten Bedingungen ermöglicht, dass Zulassungsanträge für Generika und Biosimilars bereits sechs Jahre nach Beginn des Unterlagenschutzes des Referenzarzneimittels validiert und geprüft werden dürfen. Die Zulassung selbst kann jedoch erst nach Ablauf des vollständigen Schutzzeitraums von acht Jahren erteilt werden.
Die Verpflichtungen zur Vermeidung von Lieferengpässen werden ausgeweitet. Von einer allgemeingültigen Pflicht zur Erstellung von Shortage Prevention Plans (SPP) für alle Arzneimittel wurde abgesehen. Sie gilt jedoch für sämtliche verschreibungspflichtigen Arzneimittel, Art. 117 VO-E. Unternehmen müssen drohende Engpässe mindestens sechs Monate im Voraus melden (Art. 116 VO-E). Insbesondere für mittelständische Unternehmen dürfte dies einen erheblichen Mehraufwand bedeuten.
VII. Die nächsten Schritte
Am 18. März hat der Ausschuss für öffentliche Gesundheit (SANT) im Parlament positiv über das Ergebnis der Trilog-Verhandlungen abgestimmt. Damit ist der Weg geebnet für die Übersetzung der Gesetzestexte in alle Unionssprachen und die formale Billigung durch Rat und Parlament, die noch in diesem Herbst erfolgen könnte.
Die neue Verordnung tritt 20 Tage nach Veröffentlichung im Amtsblatt der EU in Kraft, Anwendungsbeginn ist grundsätzlich 24 Monate nach Inkrafttreten. Eine unmittelbare Anwendung beziehungsweise eine kürzere Frist ist nur für wenige Ausnahmen vorgesehen, etwa die Bestimmungen zu Orphan Drugs, TEVs und Regulatory Sandboxes. Für die neue Richtlinie gilt eine Umsetzungsfrist von 24 Monaten. Ergänzend sind delegierte Rechtsakte und Leitlinien zu erwarten. Diese werden erheblichen Einfluss darauf haben, ob die Reform im Ergebnis die mit ihr verfolgten Ziele erreicht.